
H&E Basics Part 4: Troubleshooting H&E

Though the H&E stain is a relatively simple stain to perform, there are a variety of artifacts that can interfere with a good stain. Artifacts can be attributed to a variety of causes.
Sectioning often plays an essential role to achieve good quality stains. Thick and thin sections, chatter, "exploding" tissues, and floaters from the water bath are all artifacts of sectioning that may negatively impact how the tissue picks up the stain.
Thick and thin sectioning is most often a result of poor technique at the cutting station due to uneven rotation of the microtome. Many laboratories now use automated sectioning on microtomes to not only reduce ergonomic injuries, but to provide consistently even sectioned slides.

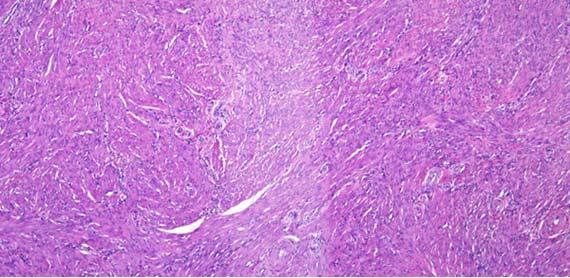
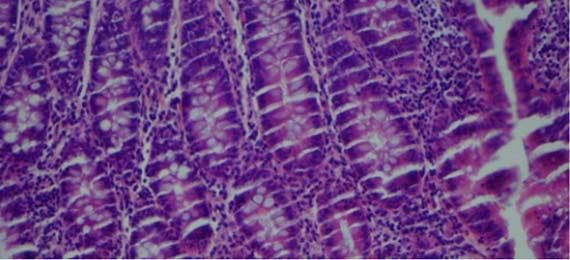
Chatter and exploding sections are typically related to tissue processing. Chatter is a result of over-processed/overly dehydrated tissue and "exploding" sections result from under-processed/poorly infiltrated tissues. Another indicator for overly processed tissues can be easily noted when looking at the red blood cells (RBCs) in vessels.
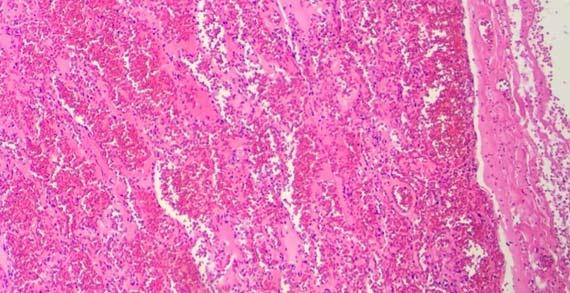
Water is often used as a differentiator for eosin, as it can gently remove excess eosin from the slide. Dehydrating alcohols and the first 100% alcohol can contain enough water to cause lightening of eosin and further differentiation of eosin. Too many "water or diluted alcohol" steps will result in lighter shades of eosin and understaining of cytoplasmic structures.
While staining, water can get into the alcohols (following eosin) due to carryover between steps. When reagents are not changed regularly, the water content will continue to increase and will be transferred to the xylenes that follow before coverslipping. This excess water in the xylene can, over time, cause seeping of the eosin from the tissue. It is seen on the slide as a pink haze. This artifact can occur even if the xylene is not visibly contaminated with water.
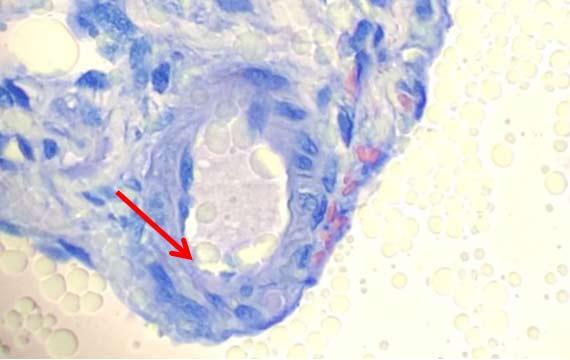
Carryover is not the only means of getting water into the xylenes. Ambient humidity can cause changes in how quickly the xylene becomes contaminated with water.
Depending on geographical location, water quality can be quite variable. Fluorination, pH, or other minerals present can impact not only how the dyes are picked up by the tissue, but also the life of the dyes. Because hematoxylin, for example, is slightly acidic, a basic pH tap water can raise the pH of the hematoxylin, making it less effective. If the tap water quality is poor or variable, the use of deionized (DI) water is a great option. Just remember to make sure the DI water pressure is sufficient for the platform stainer.
Frozen tissues can be very challenging samples to stain. Timing is so important, and teams are consistently asked to shorten stain times, allowing the pathologist to respond quickly to the surgical staff. That said, it can be difficult to find the balance between quality staining and speed.
One of the biggest artifacts I have experienced is uneven staining unrelated to frozen tissue sectioning. The number one reason is rushing the fixation and rinsing of the slide prior to staining. Remember that the media used to support frozen tissues during sectioning is water soluble and MUST be removed from the sample, not unlike removing paraffin from routine samples before staining. The media can prevent even dye infiltration just like paraffin wax does. And, not unlike wax, you will probably not see any residual media left on the slide at the end of staining, because steps during staining will remove the media along the way.
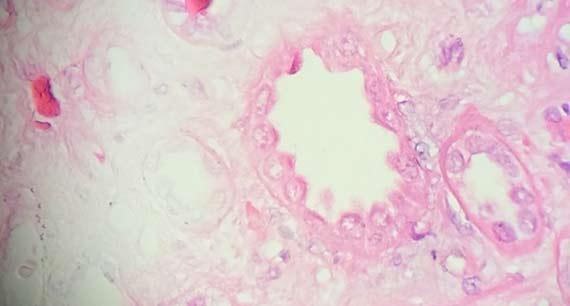
Samples are not usually collected by laboratory staff, so educating the personnel collecting samples is essential. Collecting samples using gauze pads, paper towels, cotton tipped applicators, or other intermediary devices can dry samples by the simple transfer of water from the tissue to the device. This artifact is most commonly seen with biopsies, especially when multiple samples are being collected from the same patient using the same collection device. For these cases, where rinsing the collection device with formalin could harm the patient, non-porous materials should be used to remove the tissue from the device. Saline may also be used to rinse the tissue from the device into another container, as it will not cause water to be removed from the collected sample.
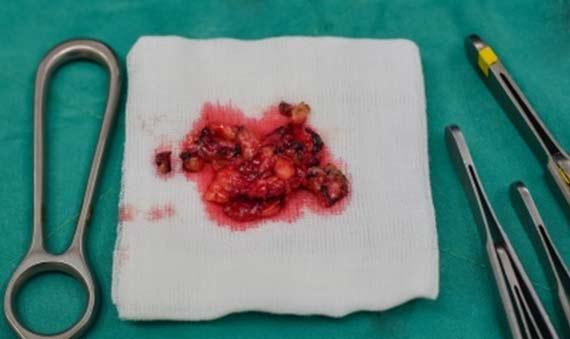
Samples should be collected and immediately immersed in fixative to ensure proper fixation. Formalin is the most commonly used fixative in the laboratory and does provide good nuclear detail. However, formalin should be treated as would any other reagent, specifically regarding storage and expiration dates. Leaving formalin in direct sunlight can alter the pH of the solution, causing an acidic environment for tissues. Acidity in fixatives tends to have two primary impacts on tissues:
- Searing of the outer edges of the tissues
- Over-drying of the tissues
Searing occurs when the outer edge of the sample had been altered to give a "burned" appearance. This change also prevents proper infiltration of reagent into the sample because the proteins along the outer portion of the sample create a barrier for reagent to enter and for water to leave.
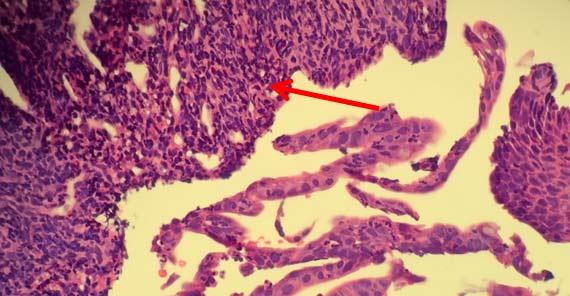
Depending on the level of acidity, the tissues can merely become overly dry. Whenever concerns about formalin viability arise, it is best to start with fresh reagent to reduce the risk of tissue damage.
As tissue processors continue to evolve, so must our processing techniques. Processing protocol times are becoming more streamlined to meet the needs of patients by creating efficiencies in processing. For this reason, putting all samples together on the same protocol is no longer reasonable. Small samples placed on an overnight protocol will become over dehydrated, making sectioning difficult. Soaking may help, but often the tissue will show excessive cracking. Large samples placed on a protocol more appropriate for biopsies will be under dehydrated and will be unlikely to be sectioned until appropriate processing has been completed.
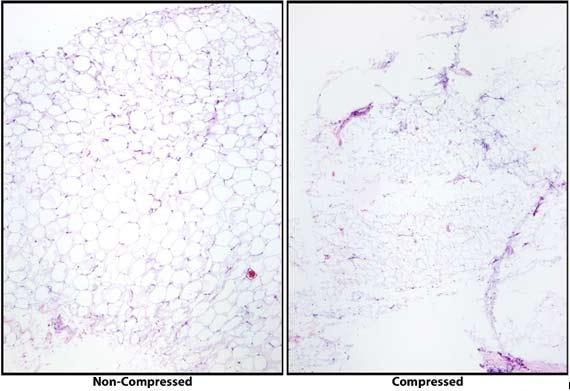
Nuclear bubbling occurs when the proteins in the nucleus coagulate. This artifact is often a result of poorly fixed samples that have encountered a high level of heat. A high pH fixative can also cause bubbling to be seen. The harsh environment cause proteins to coagulate around small droplets of liquid, which is what gives them the soap bubble appearance. Unfortunately, there is no way to remove this artifact once it has occurred.
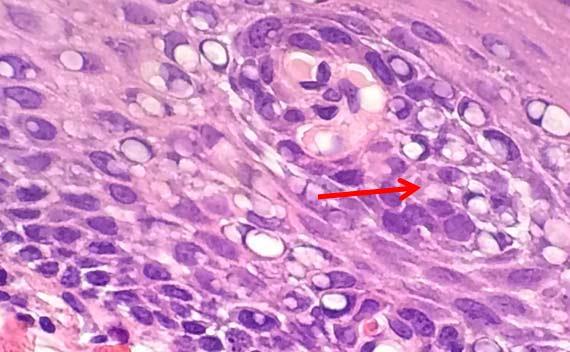
After sectioning, wet slides are often placed in an oven to dry. High temperatures (e.g., 70°C) can cause the water under the section to rapidly evaporate through the tissue. The evaporation can cause the proteins to coagulate, giving the soap bubble artifact. The best way to avoid bubbling in this case is to either lower the oven temperature or allow the slides to somewhat air dry prior to placing them in the oven.
Floaters are unfortunately all too common. The key to determining the source of the floaters is to see where they are on the slide in relation to the tissue. If they are in the same plane of focus as the tissue sample, then they most likely were a contaminate in the block (from either grossing or embedding).
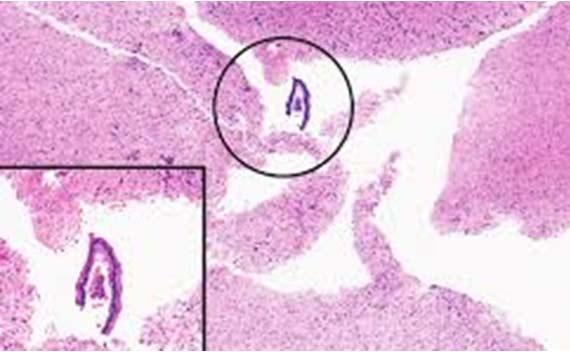
External contamination (e.g., water baths or within the staining reagents) typically will show floaters either above the tissue or out of the tissue plane. Either way, maintaining clean grossing and embedding areas, using a clean water bath, and frequently changing or filtering reagents are the best means of avoiding floaters.
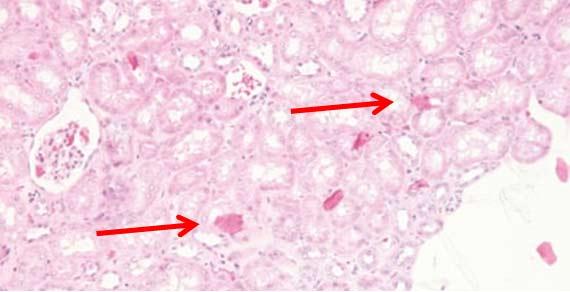
Bacteria and fungus can grow in stainers and reagent vessels if these areas are not regularly cleaned. Follow the manufacturer instructions for instrument maintenance. Remember that while bleach is appropriate for most things, it may cause issues with your instrument.
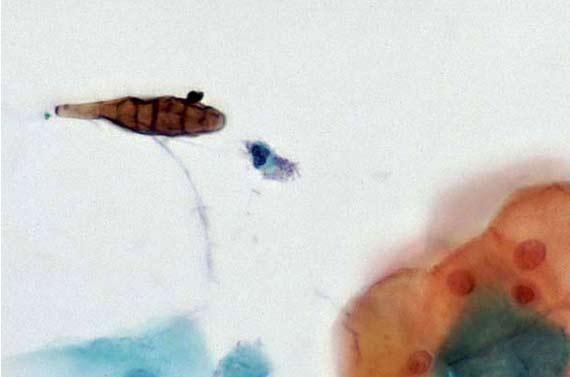
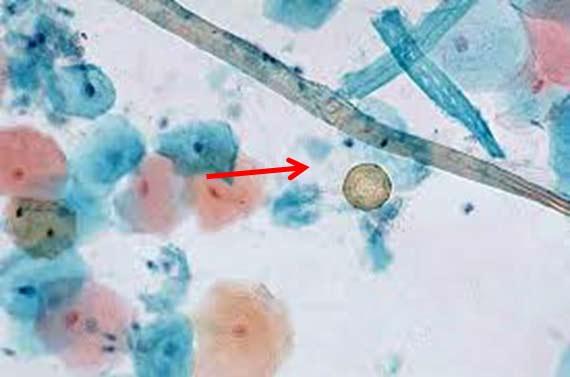
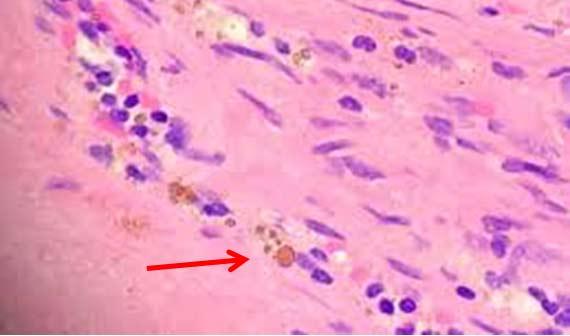
Keep in mind that the body can and does produce materials that can show up on a H&E stains as pigment. Melanin, hemosiderin (old red blood cells), and inflammatory debris can be distracting. Even fixatives can create extra cellular pigments, such as Zenker's, which can be concerning if the pathologist is not aware that the sample was fixed in something other than formalin. While Zenker's (potassium dichromate/mercuric chloride) is largely banned due to its toxicity, there are some places that may still make their own. Make a note of any unusual fixation circumstances that could alter staining.
Histology laboratories have historically used all sorts of techniques to adhere tissues to slides. In the past, when charged slides were considered too costly for routine sectioning, laboratories relied on albumin or other proteins to help tissues adhere to slides. Commercial reagents were (and still are) available to be used as additives to the water bath. The challenge, though, is consistency. Each technologist will fill their water bath differently and add different amounts of adhesive to the bath. While one person may be generous with the adhesive, another may be stingy, making background staining difficult to control.
Adhesives do have their own limitations (eg, background staining on H&E) and may even interfere with immunohistochemistry stains.
Charged slides are now readily available and work well with laboratory stains, decreasing the probability of background or other anomalies associated with adhesives. Because all slides are treated consistently, there is limited variability between slides.
Having both charged (plus) slides and adhesivesmeans using them together should make the tissue stick even better, right? Wrong! The nature of both methods is to attract ions from the tissue. When used together, the properties of both cancel each other out, essentially changing the positively charged slide into a plain glass slide.
Xylene substitutes are now widely available and can provide an additional layer of safety to the laboratory. Keep in mind that they do come with their own set of limitations, such as a low tolerance for water. It may also be necessary to use a compatible mounting media as recommended by the manufacturer, as not all of them are compatible with typical xylene-based mountants.
Although xylene substitutes are generally safer, they should still be treated with the same level of caution as xylene. Waste disposal laws vary by region, but xylene substitutes should ALWAYS be disposed of in the same manner as xylene and not poured down the drain. These reagents are still hydrocarbons that are not good for the environment. They are also typically quite aromatic, so laboratories that use substitutes tend to need somewhat better ventilation to combat the smell.
There are other alcohols on the market that can work just was well for staining as ethanol. Methanol, isopropyl alcohol (IPA), and Flex are the most common and can be more cost effective for laboratories than using ethanol. Flex alcohol manufacturers combine ethanol, methanol, and IPA together in different mixtures, so be sure to get the content percentages (if possible). If there are any concerns about using a particular reagent on a stainer, always check with the instrument manufacturer for compatibility concerns. In the end, just make sure that the stain is internally validated.
One of the easiest ways to keep H&E slides looking good is to simply ensure frequent reagent changes. Most manufacturers have some information about the number of slides that their reagents may tolerate before they must be changed. While this data is a guideline, see what makes sense for your laboratory. If changing reagents daily is a better fit, then change daily.
Most laboratories today are using some kind of staining platform. Not only does this free uptechs to do other things, it also lends itself to stain consistency. But, like all other instruments in the laboratory, maintenance and quality control are imperative to stain success. Clean and maintain your stainer per manufacturer specifications.
If laboratory is high volume, it may be appropriate to decrease the time between maintenance tasks. Either way, ALWAYS have a trained engineer perform mechanical work on the instrument. Most companies offer service plans that allow users to call in for service, and they typically will also schedule the preventative maintenance. Be aware of companies that attempt to provide service without showing their credentials for the instrument! Poor service from an outside vendor can be just as damaging as not having any service at all.
While this one seems straight forward, it is surprising just how many teams spend little time training staff on their instruments. Having regular competency checks and providing opportunities for team members to refresh their craft through webinars or other external training is a great way to make sure everyone understands the fundamentals of staining.
Of course, getting everyone to agree on what is the best stain can be a challenge. It may be easiest to program whatever staining is being used to accommodate several protocols with slightly different times, so that the pathologists have some options. I also create a simple code system so that I know what the stain times are, but the pathologists will have to respond based only on what they see.
When adjusting coloration, it is best to keep it simple. The following adjustments often work well:
- Hematoxylin +/- 30 seconds
- Eosin Y +/- 15 seconds
Typically, it is helpful to only make one adjustment at a time. These times are just enough to make subtle changes. You can be more aggressive if needed. If the hematoxylin is way too light, then certainly increase the time by one minute or more. For tweaking, however, the times above tend to work well.
REMEMBER: Sometimes simply adjusting either the hematoxylin or eosin may be enough to make the other brighter or lighter, depending on what you are looking for.
About the presenter
Cindy Sampias is a board certified Cyto- and Histo-technologist. With more than 25 years of experience, she is a guest speaker at histology and cytology meetings around the country. She is a technical author for Media Lab, publishing a variety of technical courses and sharing best practices in histology.
Related Content




Leica Biosystems Knowledge Pathway content is subject to the Leica Biosystems website terms of use, available at: Legal Notice. The content, including webinars, training presentations and related materials is intended to provide general information regarding particular subjects of interest to health care professionals and is not intended to be, and should not be construed as, medical, regulatory or legal advice. The views and opinions expressed in any third-party content reflect the personal views and opinions of the speaker(s)/author(s) and do not necessarily represent or reflect the views or opinions of Leica Biosystems, its employees or agents. Any links contained in the content which provides access to third party resources or content is provided for convenience only.
For the use of any product, the applicable product documentation, including information guides, inserts and operation manuals should be consulted.
Copyright © 2026 Leica Biosystems division of Leica Microsystems, Inc. and its Leica Biosystems affiliates. All rights reserved. LEICA and the Leica Logo are registered trademarks of Leica Microsystems IR GmbH.