
Freezing Biological Samples

Neuroscience researchers usually need to see slices of whole brain in order to determine the location in brain of detail they are viewing under the microscope. The brain needs to be hardened to allow cutting the thin slices, unless a vibrating blade microtome, which is much slower, is used. Paraffin embedding as commonly used for medical samples works only for small or thin samples, paraffin will not penetrate to the center of a whole rodent brain. Thus, neuroscience researchers usually use freezing to harden the brain tissue for sectioning.
Three solid forms of water
Freezing damages cell membranes, and reduces the histological readability of biological specimens. This is a discussion of why, the consequences, and how to minimize or avoid the damage when using freezing as the means to harden biological tissue in order to cut thin sections for histology.
Pure water can exist in the solid state in three forms. There are two crystalline forms, hexagonal crystal, and cubic crystal. Water can also be frozen, so rapidly it does not have time to form crystals, and remains amorphous (vitreous form). Crystal formation is responsible for the expansion of water as it freezes (expansion on freezing is a property unique to water), so vitreous ice does not expand upon solidification. This makes it the only desirable form of freezing for biological specimens.
Rate of freezing critical
Expansion of the water content by freezing stretches and penetrates cell membranes. Slow freezing promotes ice crystal formation, and thus expansion. Biological materials are poor thermal conductors. It is therefore likely that thermal gradients will exist as the sample is frozen, and some ice crystals will form in the interior of any piece of tissue over 10 mm from the cold source. Placed on a cold surface to freeze, specimens will freeze slowly and will thus likely form crystals, especially in parts most distal from the freezing surface. When sectioned, slowly frozen tissue with large crystal formation will have the well known “swiss cheese” effect, multiple holes and lost cell contents, to varying degrees, depending on the degree and size of crystal formation.
Note that it is the rate of freezing, not the end-temperature, that is critical at first. Fast freezing depends on the percentage of the total surface in contact with the cold source, the volume of water, the starting temperature of the water (specimen), and how cold the source is. Ideally, the cold source is at an extremely cold temperature and arranged to contact all surfaces.
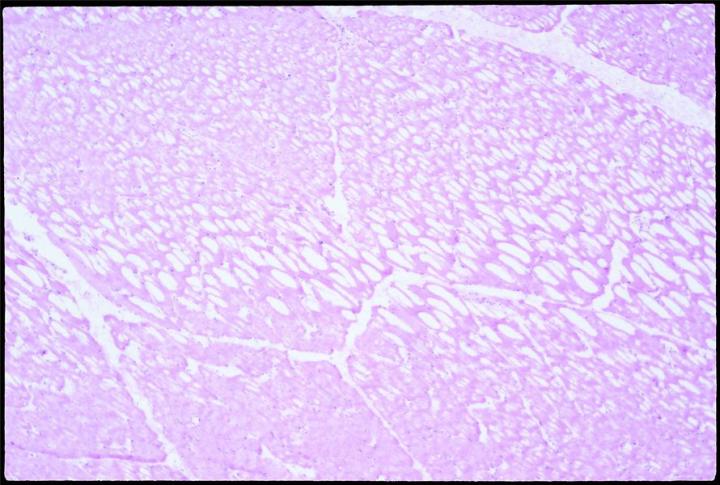
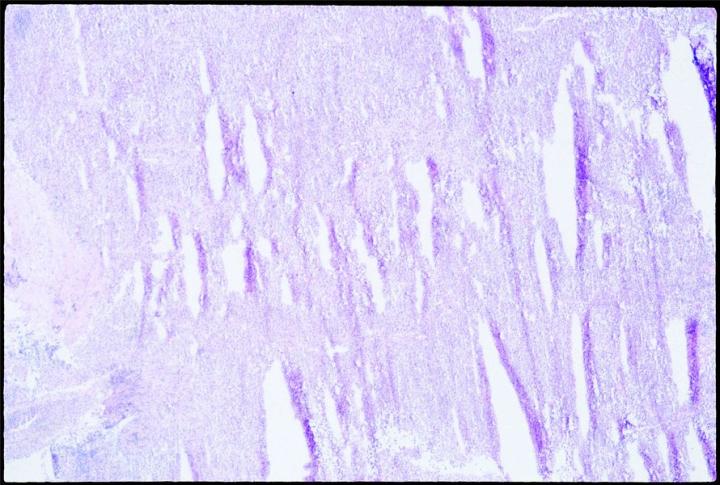

Sectioning as soon as possible
However, even if a specimen is successfully quick frozen as vitreous, it may not stay that way. Vitreous ice is an unstable state. At above -121˚C, amorphous ice begins to gradually restructure as cubic ice, and expand. At above -80˚C, cubic ice begins to restructure as hexagonal ice, and expands again. Fortunately, this is a slow conversion process, depending on temperature gradients, and purity of the water.
In most common applications, tissue must be warmed above the snap freezing temperature in order to section it (-9 to -19˚C for brain, but depending on the tissue type). Otherwise, it is too cold to cut, and shatters when bent by the knife bevel. The visible symptom of cutting too cold is “shutters” or shatter marks parallel to the knife blade. The sectioning should begin as soon as the tissue is warm enough to cut. Tissue should never be left overnight or for long periods in a cryostat, even if there is no defrost cycle. The conversion from vitreous ice to large hexagonal crystals will begin. Crystals will expand the ice, and damage cells.
Liquid nitrogen
To freeze most quickly, immersion in a liquid creates the most surface contact. An acceptable second is to place on a prechilled metal pedestal, and quickly surround the tissue with powdered dry ice, powdered to maximize surface contact.
Liquid nitrogen is one of the coldest liquids routinely available. It does not mix with tissue. These properties would seem to make it an ideal freezing medium. However, liquid nitrogen has an extremely low specific heat constant. The result is that it boils locally and copiously at the contact point with even a small piece of warmer tissue. One consequence is that a vapor barrier, a layer of nitrogen gas, can build up next to the tissue, and insulate and greatly slow penetration of cold into the tissue, and thus slow the rate of freezing in an unpredictable pattern. As a result, it often happens that the outer layer of tissue is quick frozen and vitreous, but then the vapor barrier is formed, cold penetrates more slowly, and the inner part freezes slower. Crystal formation expands the tissue inside the frozen shell, and the entire block cracks. A cracked block indicates that at least the innermost tissue has been frozen crystalline. Note that this is a problem with large blocks of tissue (for example, a whole rat or even mouse brain), but liquid nitrogen works very well to freeze biopsy or smaller samples, and is used successfully in clinical histology labs.
Other solutions
Various other liquids are used more reliably for larger blocks, most notably isopentane cooled to -80˚C. It does not form vapor barriers, and if the tissue is little more than a cubic centimeter, it will freeze as all vitreous. Full immersion in the liquid is necessary for maximum surface contact, and harmless to the tissue. Movement through the liquid is helpful.
Another solution is to permeate or perfuse the tissue with glycerine, polyethylene glycol (antifreeze), glycerol, or sucrose solutions. These will disrupt crystal formation, and lower the freezing temperature. Sucrose at 30% in phosphate buffer is commonly used, but only with well fixed tissue, after fixation, as the osmolarity would shrivel unfixed tissue.
About the presenter
References
- Jongebloed, W.L., Stokroos, D., Kalicharan, D., and Van der Want, J.J.L. Is Cryopreservation Superior Over Tannic Acid/Arginine/Osmium Tetroxide non-Coating Preparation in Field Emission Scanning Electron Microscopy. Scanning Microscopy 13: 93-109, 1999.
- Reid N. and Beesley J. Sectioning and Cryosectioning for Electron Microscopy. Practical Methods in Electron Microscopy, Vol 13, Elsevier, Amsterdam
Related Content




Leica Biosystems Knowledge Pathway content is subject to the Leica Biosystems website terms of use, available at: Legal Notice. The content, including webinars, training presentations and related materials is intended to provide general information regarding particular subjects of interest to health care professionals and is not intended to be, and should not be construed as, medical, regulatory or legal advice. The views and opinions expressed in any third-party content reflect the personal views and opinions of the speaker(s)/author(s) and do not necessarily represent or reflect the views or opinions of Leica Biosystems, its employees or agents. Any links contained in the content which provides access to third party resources or content is provided for convenience only.
For the use of any product, the applicable product documentation, including information guides, inserts and operation manuals should be consulted.
Copyright © 2026 Leica Biosystems division of Leica Microsystems, Inc. and its Leica Biosystems affiliates. All rights reserved. LEICA and the Leica Logo are registered trademarks of Leica Microsystems IR GmbH.